
▲第一作者:杨梦莹
共同通讯作者:吕湘龙、唐鑫
通讯单位:桂林理工大学材料科学与工程学院,有色金属与材料加工新技术教育部重点实验室,有色金属矿床勘查与资源高效利用协同创新中心
论文DOI:10.1021/acscatal.4c00217 (点击文末「阅读原文」,直达链接)
采用氧化衍生方法(Oxide-Derived, OD)制备的纳米多孔金属在CO2电化学还原中往往具有优异的催化性能。本文采用一种简单、经济的电化学氧化还原方法制备得到了OD-Ag电极,并研究了其在长时间CO2电催化过程中的结构演变。电子背散射衍射(EBSD)分析表明,在初始CO2电化学还原阶段(前30 min),银表面氧化物还原生成OD-Ag的过程中存在明显的晶粒细化现象。实验结果表明催化活性和选择性与电极表面的晶界密度和纳米多孔结构密切相关。长时间催化反应(12 h)后,表面结构发生明显变化,表面晶粒粗化,纳米结构层厚度减小,导致OD-Ag电极活性降低。文中提出了一种催化过程中溶解/再沉积的机制解释了OD-Ag电极失活的原因,并且通过对失活电极再次电化学氧化还原处理,可使其纳米多孔层厚度和电化学活性面积显著增加,从而使催化活性再次恢复。
大量研究表明由相应氧化物衍生(oxide-derived metals, OD)的金属催化剂可以显著提高电化学二氧化碳还原的催化活性和选择性。但是目前对其高活性来源和性能增强的机理仍存在较大争议。此外,在长时间电催化过程中,催化剂的表面结构和成分经常会由于表面重构而发生变化,导致催化性能降低。这也给研究催化剂的表面结构和性能关系带来了更大的挑战和困难。
斯坦福大学Kanan课题组成功制备了OD Au、OD Cu和Sn/SnOx复合材料作为CO2电化学还原的催化剂,通过定量表征CO2催化活性与晶界密度的关系,以及采用扫描探针直接探测晶界处的活性等方法,提出了晶界活性位点理论。荷兰代尔夫特理工大学W.A. Smith课题组报道了由氧化衍生制备的纳米多孔银电极(OD-Ag),他们认为其高活性来自于电极的表面结构,因为其不仅能提高中间产物*COOH的稳定性,还能在催化剂表面附近产生高局域pH值,有助于CO2还原和抑制析氢。通过现场原位扩展X射线吸收精细结构研究,他们证明OD Ag电极催化活性提高可能来自于表面微量的氧化物和纳米结构导致的高局域pH值。此外,其他一些研究也认为,OD催化剂的本征活性受到表面形貌、表面氧含量、晶格缺陷或高指数晶面的影响。这些研究表明,目前对OD金属催化剂的高活性来源和性能增强的机理仍存在较大争议。而且在长时间电催化过程中,催化剂表面的结构和成分经常会由于表面重构而发生动态变化,导致催化性能降低。如何延长催化剂的使用寿命,使催化剂能保持长时间反应活性和稳定性是电催化CO2还原领域的热点问题。
1. 本文系统研究了电化学氧化还原制备得到的纳米多孔银(OD-Ag)电极在长时间催化反应过程中的结构演化和活性来源。OD-Ag电极在0.1 M KHCO3溶液中-0.8 V vs. RHE下生成CO的法拉第效率能达到87%,是未经处理的银箔的4倍(~22%)。
2. EBSD和SEM实验分析表明,OD-Ag电极的高活性主要来自于电极表面的高密度晶界和纳米多孔结构。在初始CO2电化学还原阶段(前30 min),银表面氧化物还原生成OD-Ag的过程中存在明显的晶粒细化现象,丰富的晶界能提供更多的催化活性位点;二是在电化学氧化还原过程中表面形成的纳米多孔结构,增加了电化学活性面积(ECSA)。理论计算也证实表面弯曲的纳米多孔结构能更好的吸附和活化CO2分子。
3.经过长时间催化反应后,OD-Ag电极会发生明显的表面重构,表面晶粒粗化,纳米结构层厚度减小, ECSA降低,导致OD-Ag电极活性降低。本文提出了一种电极催化过程中溶解/再沉积的机制解释了OD-Ag电极失活的原因。通过对失活电极再次电化学氧化还原处理,可使其纳米多孔层厚度和电化学活性面积显著增加,从而使催化活性再次恢复。
本文通过电化学氧化还原处理将Ag foil制备成纳米多孔的OD-Ag电极的形貌如图1a-c所示。Ag foil在0.2 M NaOH电解液电化学氧化5 h后得到的Ag oxide电极表面呈颗粒状和碎片状,Ag oxide在0.1 M KHCO3电解液中电化学还原30 min后得到纳米多孔OD-Ag-1电极。Ag foil、Ag oxide和OD-Ag-1的晶粒尺寸变化如图1d-f所示,经过CO2还原后电极表面晶粒发生明显细化。这种表面形貌的剧烈变化表明,OD-Ag电极在初始CO2还原电解过程中存在明显的表面重构。TEM表征证实了OD-Ag-1中的高密度晶界,这与EBSD结果一致。XRD和XPS测试表明,OD-Ag-1在CO2还原后电极表面主要成分为金属银单质。
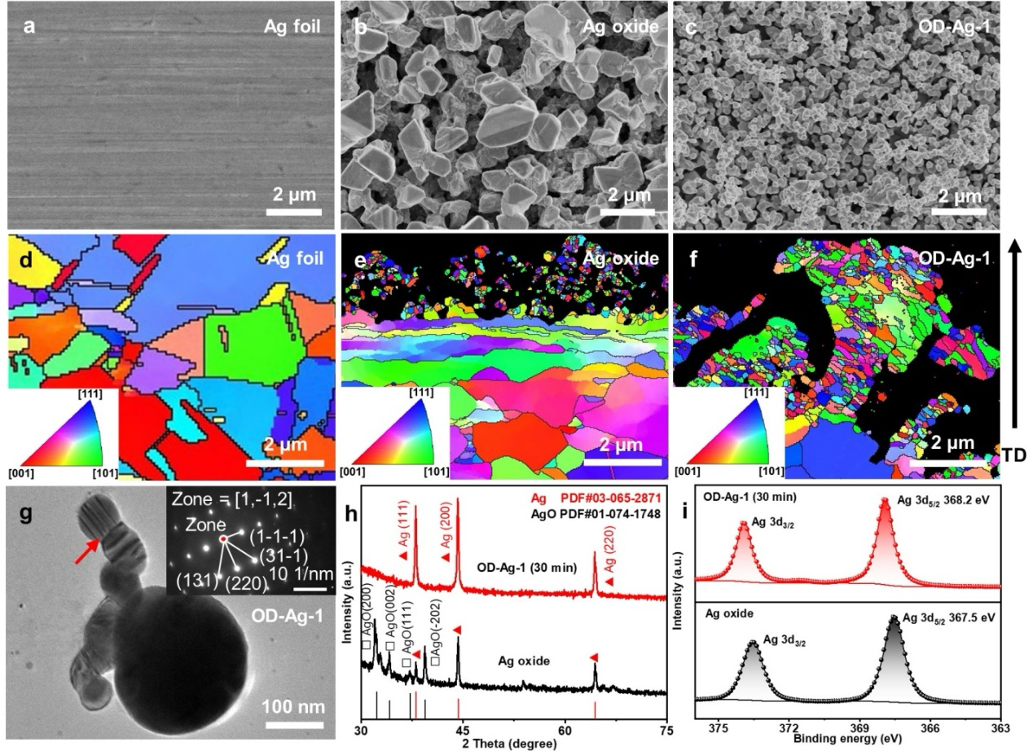
图1 Ag foil, Ag oxide和OD-Ag-1电极的微观结构表征
在CO2饱和的0.1 M KHCO3 (pH~6.83)条件下,OD-Ag电化学CO2还原成CO的法拉第效率~87%,是未经处理的Ag foil的4倍(22%)。通过线性扫描伏安曲线、电化学活性面积测试和Tafel斜率的计算可以看出OD-Ag电极相较于Ag foil电极而言具有更低的过电位、更大的电化学活性面积和更快的电子转移速率,这证明OD-Ag具有较高的CO2还原催化活性。
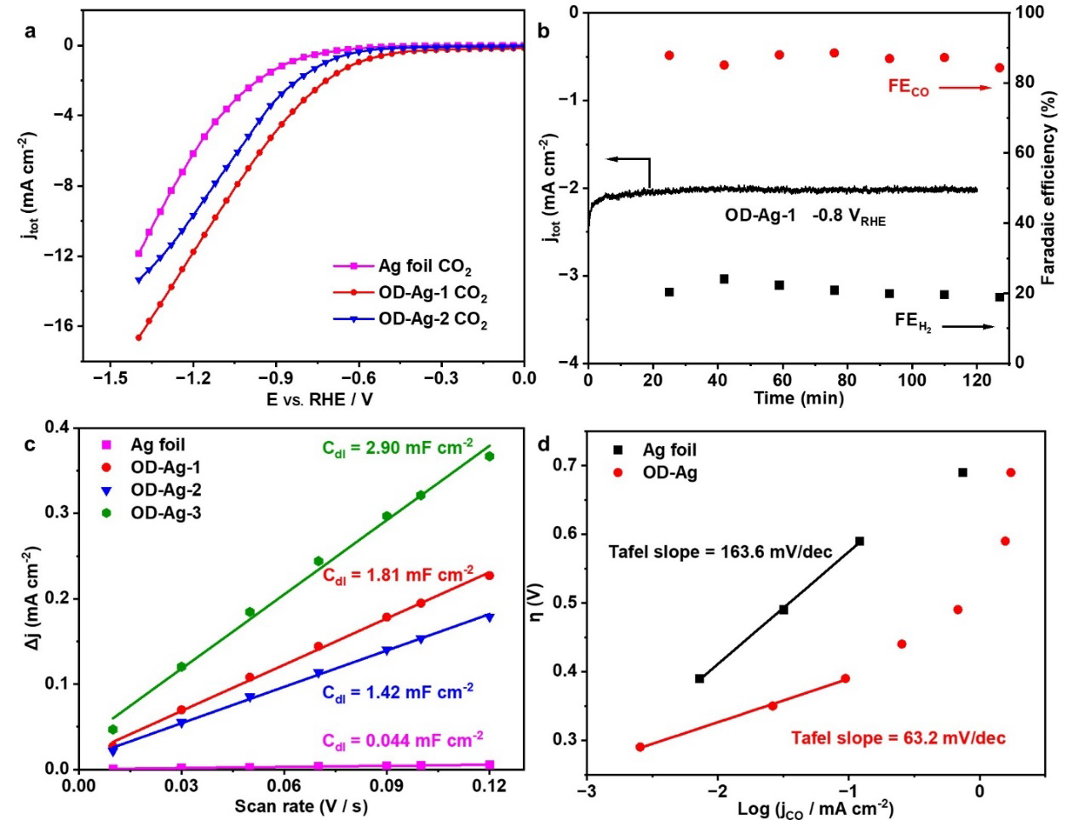
图2 Ag foil、OD-Ag-1 (30 min)和OD-Ag-2 (12 h)电极CO2电催化性能测试
图3为Ag-foil和Porous-Ag的理论计算模型。通过对OD-Ag催化活性进行理论计算发现,具有相同(111)晶面的Porous-Ag由于表面的台阶结构会使得C-O键弯曲和键长拉长,从而有利于在外部边缘位点处对CO2进行吸附和活化。晶体轨道哈密顿布居(COHP)计算也证实Porous-Ag外边缘位点的C-O键具有更高的ICOHP积分值(-3.53 eV),因此更容易断裂C-O键,从而促进CO2的活化行为。反应路径分析发现,在Ag-foil上的速度控制步骤为CO2的活化步骤(*+CO2 to *CO2),而在Porous-Ag上的速度控制步骤是质子耦合电子转移(PCET)过程(*CO2 to *COOH)。这可能是由于在高度弯曲的Porous-Ag表面存在大量低配位的Ag原子,能有效降低CO2活化步骤的活化能。
图3 Ag-foil和porous-Ag的理论计算和分析
为了测量OD-Ag-1的稳定性,在-0.8 V电位下0.1 M KHCO3电解液中进行了长达12 h的CO2电化学还原。如图4a所示,在12 h的电解过程中,总电流密度从2.55 mA cm-2缓慢下降到1.78 mA cm-2,FECO值从87%显著下降到69%,表明CO2还原催化活性严重下降。同时,值从13%增加到31%,表明随着反应时间的增加,析氢反应加剧。通过SEM和EBSD测试发现,经过12 h的CO2还原后,OD-Ag的纳米多孔结构韧带粗化,晶粒尺寸增加,纳米多孔层的厚度减少(从8.7 mm降低到7.2 mm)。ECSA测试发现OD-Ag-2 (12 h)的电化学活性面积也显著减少(图2c)。这些反应过程中的结构变化导致了OD-Ag电极的CO2电催化活性随时间逐渐降低。
图4 OD-Ag电极的长时间稳定性测试以及测试后的结构表征
文中作者提出了一种电极催化过程中溶解/再沉积的机制来解释OD-Ag电极长时间反应失活的原因。如图5所示,在CO2还原初期,银表面残余氧化物在还原电位作用下倾向于溶解到电解液中生成Ag离子,发生瞬时溶解;长时间反应过程中,当表面氧化物消耗殆尽后,电极表面的中间产物(如*COOH)会与表面金属原子形成金属-吸附配合物,这种金属-吸附配合物在电极表面的迁移会继续导致OD-Ag电极的溶解。通过ICP测试电解液中Ag+的浓度也证实了Ag的溶解。然而,溶液中溶解的Ag离子在还原电位作用下通常是不稳定的,部分Ag离子可能会在表面重新沉积为金属Ag,导致纳米孔的堵塞,韧带结构粗化,电化学活性面积降低,催化性能下降。
图5 OD-Ag电极在长期催化反应过程中的溶解/再沉积机制
受电化学氧化还原过程中OD-Ag表面重构的启发,我们将长时间反应活性降低的电极再次进行电化学氧化-还原处理得到电极OD-Ag-3。从图6a可以看出,OD-Ag-3的FECO值达到了~85%,并可维持至少2 h活性不衰退,而且OD-Ag-3的电流密度从1.78 mA cm-2显著增加到4.61 mA cm-2。SEM分析发现再次处理后的电极表面形成了分层状的纳米多孔结构,其韧带和孔隙较小,纳米多孔层厚度和电化学活性面积都明显增加,从而使其催化活性再次恢复。
图6 (a-b) 再活化后的OD-Ag-3电极结构和性能;(c) OD-Ag催化剂在CO2还原过程中的制备和结构演化示意图
本工作通过电化学氧化还原处理制备得到OD-Ag催化剂在电化学CO2还原中表现出较高的催化活性,电催化CO2为CO的法拉第效率约为87%,是相同条件下未处理Ag箔的4倍。OD-Ag催化活性提高主要有两个原因:一是EBSD分析表明,在初始CO2电化学还原阶段(前30 min),银表面氧化物还原生成OD-Ag的过程中存在明显的晶粒细化,丰富的晶界能提供更多的催化活性位点;二是表面形成的纳米多孔结构,显著增加了电极的电化学活性面积。经过长时间催化反应(12 h)后,表面结构会发生明显重构,表面晶粒粗化,纳米结构层厚度减小,导致OD-Ag电极催化活性降低。对失活的OD-Ag电极再次进行电化学氧化还原处理可以恢复其催化活性。
这项工作不仅为氧化衍生催化剂的结构演变过程提供了新的理解,而且还介绍了一种新的电化学方法来恢复失活电极的活性,从而以延长电催化剂在长时间催化反应中的寿命。
杨梦莹,桂林理工大学吕湘龙和唐鑫课题组硕士研究生,研究方向为纳米多孔金属制备及电化学催化。
吕湘龙,桂林理工大学副教授、硕士生导师,主要研究方向为纳米多孔金属材料的制备及电催化性能研究。在ACS Catalysis、Acta Materialia、Journal of Materials Science &Technology等SCI期刊上发表学术论文20余篇。
唐鑫,桂林理工大学教授、博士生导师,目前主要从事基于第一性原理的电催化功能材料和固体离子电解质设计及制备,以及铝合金新型焊接材料的研发工作。承担国家级项目2项,省部级项目2项,发表学术论文30余篇,授权专利2项。